Contents
- Important Notice
- Preparation
- Building pre-computed correlation data (optional)
- Computing relationships based on root-related data
- 3.1 Computing clusters
- 3.2 Generating pictures with intensity fold-changes
- Computing relationships based on all array data
- Comparing graphs
- 5.1 Via mapping
- 5.2 Via graph-level operation
- Fishing genes that are highly correlated
- Computing GO enrichment and getting gene list (optional)
This is a step-by-step running script. All data mentioned in this page are available and users can try all commands one by one. For detailed description of each program, please refer the Command line manual.
System Requirement:
- Java Environment: Be sure that you have Java Runtime Environment (JRE) installed in your computer. If not, JRE can be downloaded from java.sun.com. In case that you don't know how to download/install JRE, please contace your technical support.
- The Graphviz program is required for making pictures, and it is available at http://www.graphviz.org/.
- In practice, we found that 1.4GB memory was needed for some programs. So we suggest to use a computer with at least 2GB memory to run this tool kit.
For this example:
We need only the extracted files, the zip files are no longer required. Copy all these files:
- affy23k_1436_arrays_for_megacluster.txt
- ATH1_ProbeLocusMap.TXT
- ATH1_Root_RMA.tdt
- colorALL.txt
- genelist.txt
- geneSymbols.txt
- maccu.jar
to your working directory (in this example it is D:\wdlin\Project1\maccu\data\20091203_example).
In MACCU, we use RelationComputer and CoExpressFishing to build up graphs based on co-expression relationships, where a co-expression relationship between two genes is defined if the Pearson correlation coefficient between their expression levels is above a user-defined threshold. This means that we have to compute Pearson correlation coefficients between all pairs of involved genes. This kind of computation will be very time-consuming when we are doing fishing because the term "involved genes" for fishing means "all genes." To save time from computing these relationships, we compute them once and reuse the computation results for many times. The program ExpressionReader can do this using the following command, and this command computes Pearson correlation coefficients and save them into file ATH1_Root_RMA.correl. Note that this pre-computation step may cost a few hours to a few days.
D:\wdlin\Project1\maccu\data\20091203_example>java -Xmx1400M -classpath maccu.jar maccu.ExpressionReader -I ATH1_Root_RMA.tdt -O ATH1_Root_RMA.correl
program: ExpressionReader
input filename (-I): ATH1_Root_RMA.tdt
output filename (-O): ATH1_Root_RMA.correl
number filer (-NF): 8
correl filer (-CF): 0.5
input file with probe assignment (-assign): false
reading expression
computing correlation
progress: 0% = 100000 / 260148050
progress: 0% = 200000 / 260148050
progress: 0% = 300000 / 260148050
progress: 0% = 400000 / 260148050
(deleted)
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project1\maccu\data\20091203_example>java -Xmx1400M -classpath maccu.jar maccu.RelationComputer -I ATH1_Root_RMA.tdt -O root -P genelist.txt -assign ATH1_ProbeLocusMap.TXT -CF 0.7
program: RelationComputer
expression filename (-I): ATH1_Root_RMA.tdt
probe assignment filename (-assign): ATH1_ProbeLocusMap.TXT
correlation filename (-C): null
output prefix (-O): root
probe filename (-P): genelist.txt
number filer (-NF): 8
correl filer (-CF): 0.7
by locus (-L): true
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project1\maccu\data\20091203_example>java -Xmx1400M -classpath maccu.jar maccu.GraphAdjust -I root.graph -O root
program: GraphAdjust
input graph (-I): root.graph
output prefix (-O): root
remove node list (-RN): []
remove edge list (-RE): []
remove graph list (-remove): []
retain graph list (-retain): []
degree filter (-D): 0
cluster-size filter (-S): 1
cluster 0, size: 61
cluster 1, size: 15
cluster 2, size: 4
cluster 3, size: 2
cluster 4, size: 2
D:\wdlin\Project1\maccu\data\20091203_example>
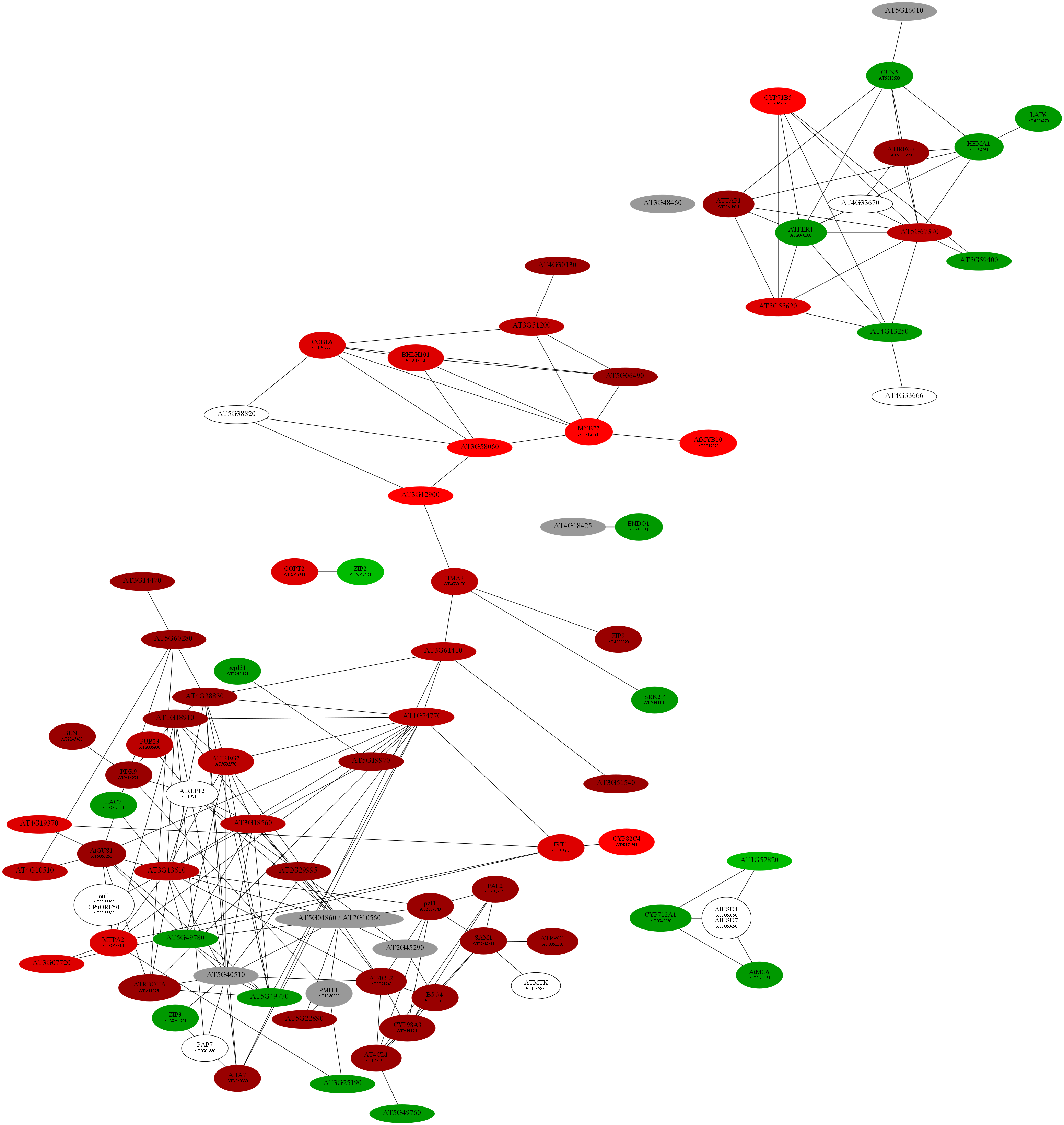
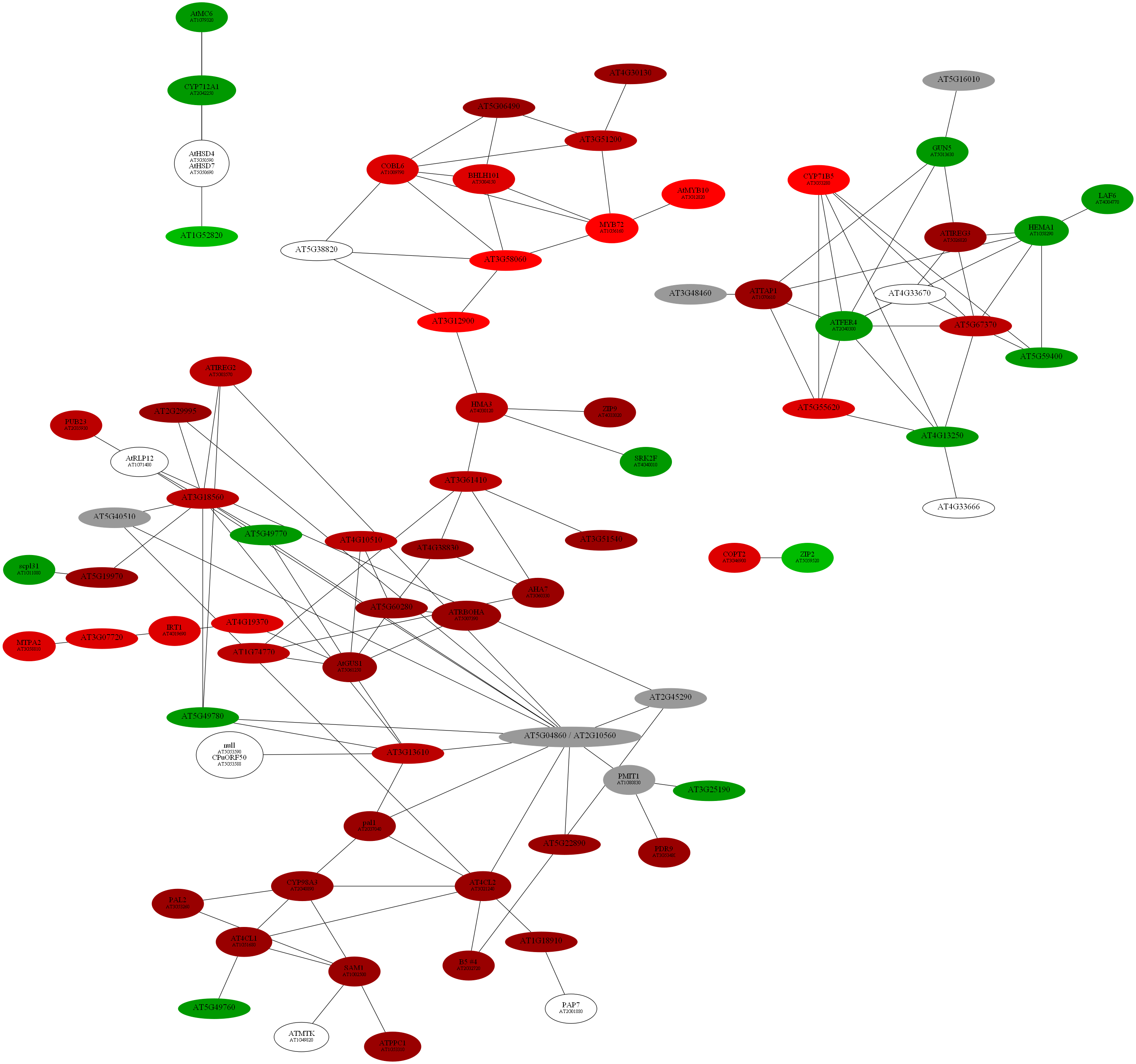
D:\wdlin\Project1\maccu\data\20091203_example>java -classpath maccu.jar maccu.GraphMakeUp -I root.graph -O root.dot -label geneSymbols.txt -fold colorALL.txt
program: GraphMakeUp
input graph (-I): root.graph
output filename (-O): root.dot
label filename (-label): geneSymbols.txt
fold change filename (-fold): colorALL.txt
coloring list (-C): {}
font 1 (-font1): 12
font 2 (-font2): 8
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project1\maccu\data\20091203_example>twopi -Goutputorder=edgesfirst -Goverlap=vpsc -Granksep=2 -Gratio=auto -Tpng -o root.png root.dot
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project1\maccu\data\20091203_example>neato -Goverlap=false -Gsplines=true -Gsep=.1 -Tpng -o root.png root.dot
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project1\maccu\data\20091203_example>java -Xmx1400M -classpath maccu.jar maccu.RelationComputer -I affy23k_1436_arrays_for_megacluster.txt -O 1436 -P genelist.txt -assign ATH1_ProbeLocusMap.TXT -CF 0.7
program: RelationComputer
expression filename (-I): affy23k_1436_arrays_for_megacluster.txt
probe assignment filename (-assign): ATH1_ProbeLocusMap.TXT
correlation filename (-C): null
output prefix (-O): 1436
probe filename (-P): genelist.txt
number filer (-NF): 8
correl filer (-CF): 0.7
by locus (-L): true
D:\wdlin\Project1\maccu\data\20091203_example>java -Xmx1400M -classpath maccu.jar maccu.GraphAdjust -I 1436.graph -O 1436
program: GraphAdjust
input graph (-I): 1436.graph
output prefix (-O): 1436
remove node list (-RN): []
remove edge list (-RE): []
remove graph list (-remove): []
retain graph list (-retain): []
degree filter (-D): 0
cluster-size filter (-S): 1
cluster 0, size: 39
cluster 1, size: 9
cluster 2, size: 4
cluster 3, size: 2
cluster 4, size: 2
cluster 5, size: 2
cluster 6, size: 2
cluster 7, size: 2
cluster 8, size: 2
cluster 9, size: 2
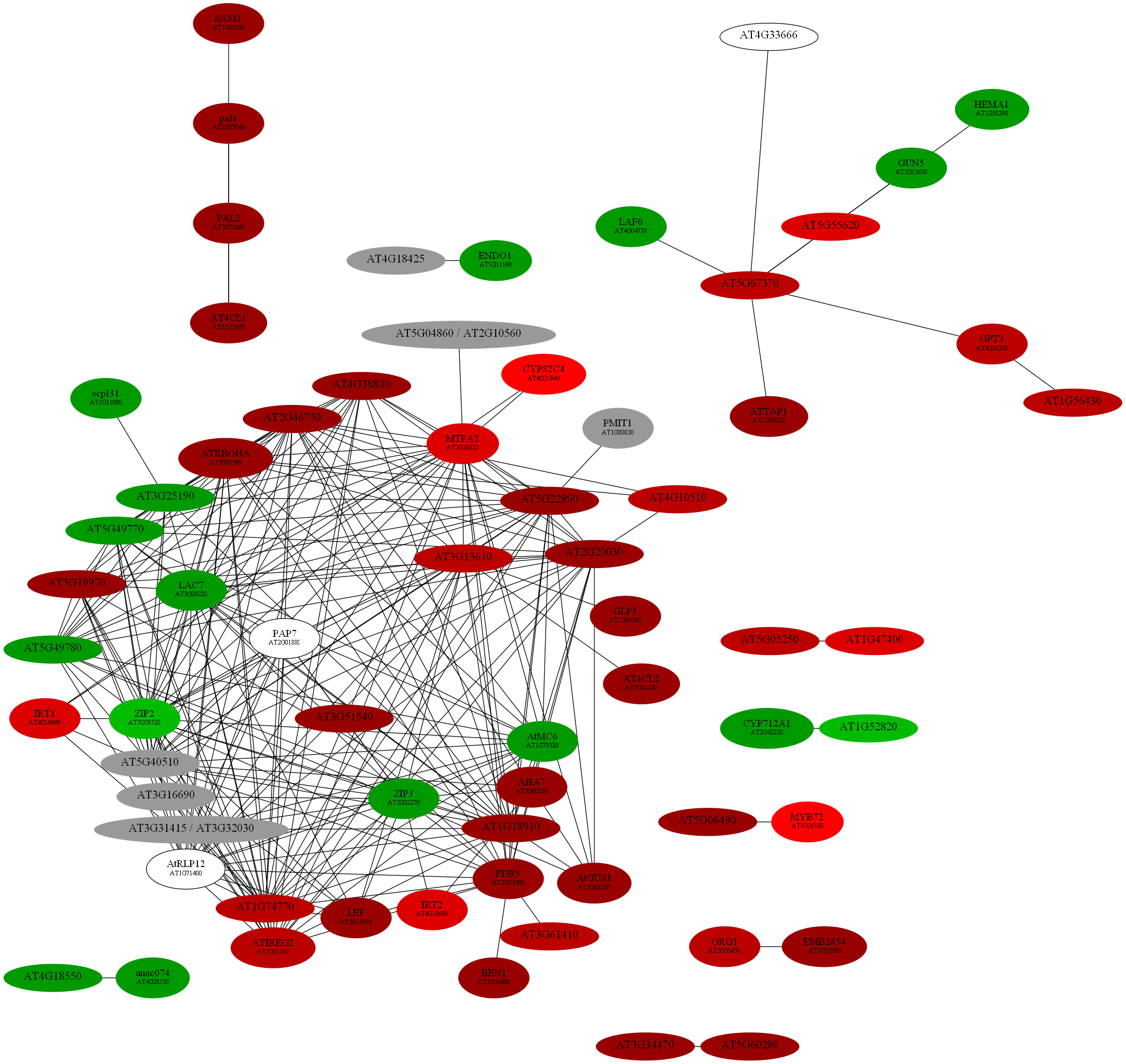
D:\wdlin\Project1\maccu\data\20091203_example>java -classpath maccu.jar maccu.GraphMakeUp -I 1436.graph -O 1436.dot -label geneSymbols.txt -fold colorALL.txt
program: GraphMakeUp
input graph (-I): 1436.graph
output filename (-O): 1436.dot
label filename (-label): geneSymbols.txt
fold change filename (-fold): colorALL.txt
coloring list (-C): {}
font 1 (-font1): 12
font 2 (-font2): 8
D:\wdlin\Project1\maccu\data\20091203_example>twopi -Goutputorder=edgesfirst -Goverlap=vpsc -Granksep=2 -Gratio=auto -Tpng -o 1436.png 1436.dot
D:\wdlin\Project1\maccu\data\20091203_example>
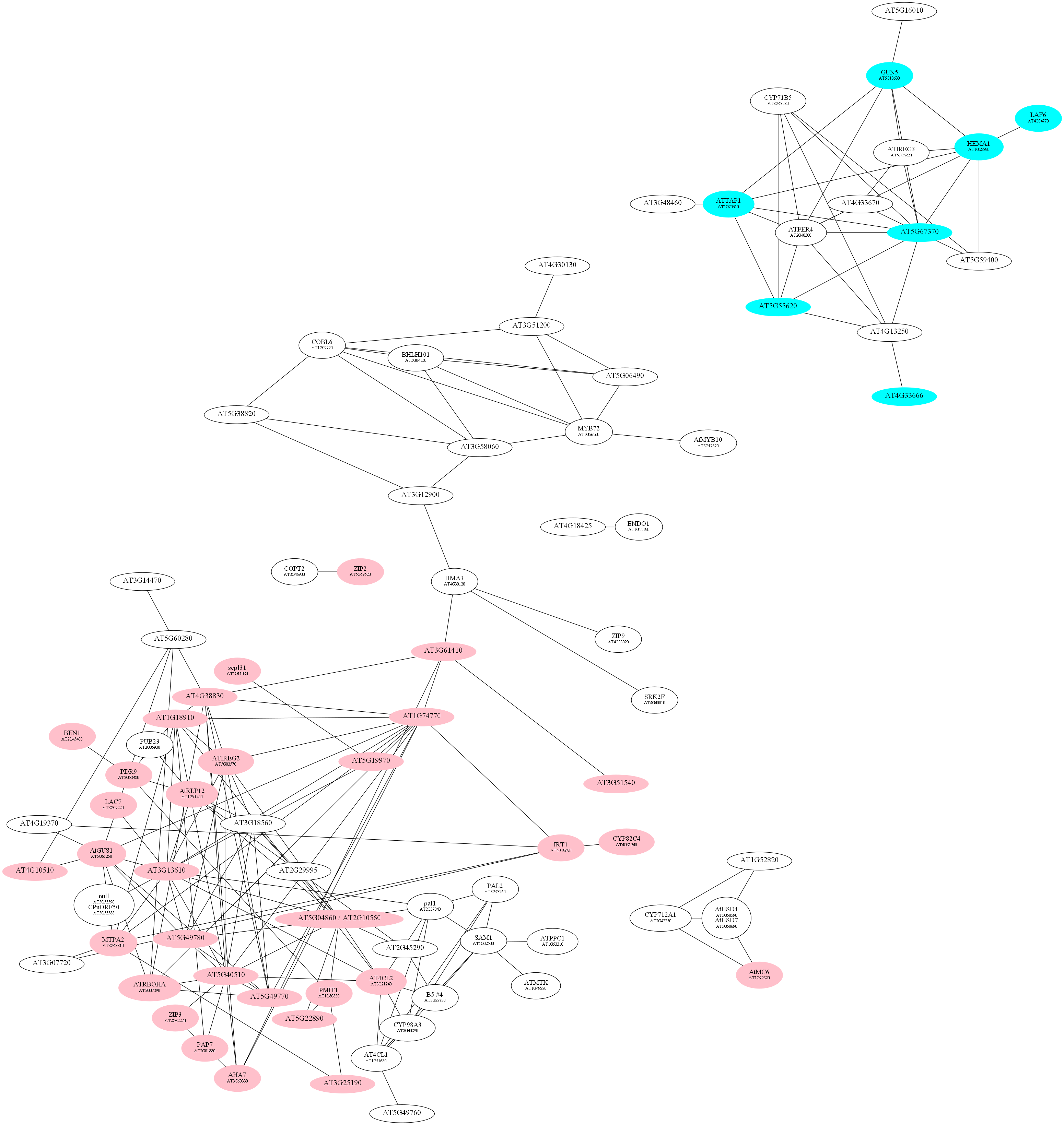
D:\wdlin\Project1\maccu\data\20091203_example>java -classpath maccu.jar maccu.GraphMakeUp -I root.graph -O root1436.dot -label geneSymbols.txt -C pink 1436-0.graph -C cyan 1436-1.graph
program: GraphMakeUp
input graph (-I): root.graph
output filename (-O): root1436.dot
label filename (-label): geneSymbols.txt
fold change filename (-fold): null
coloring list (-C): {pink=1436-0.graph, cyan=1436-1.graph}
font 1 (-font1): 12
font 2 (-font2): 8
D:\wdlin\Project1\maccu\data\20091203_example>twopi -Goutputorder=edgesfirst -Goverlap=vpsc -Granksep=2 -Gratio=auto -Tpng -o root1436.png root1436.dot
D:\wdlin\Project1\maccu\data\20091203_example>
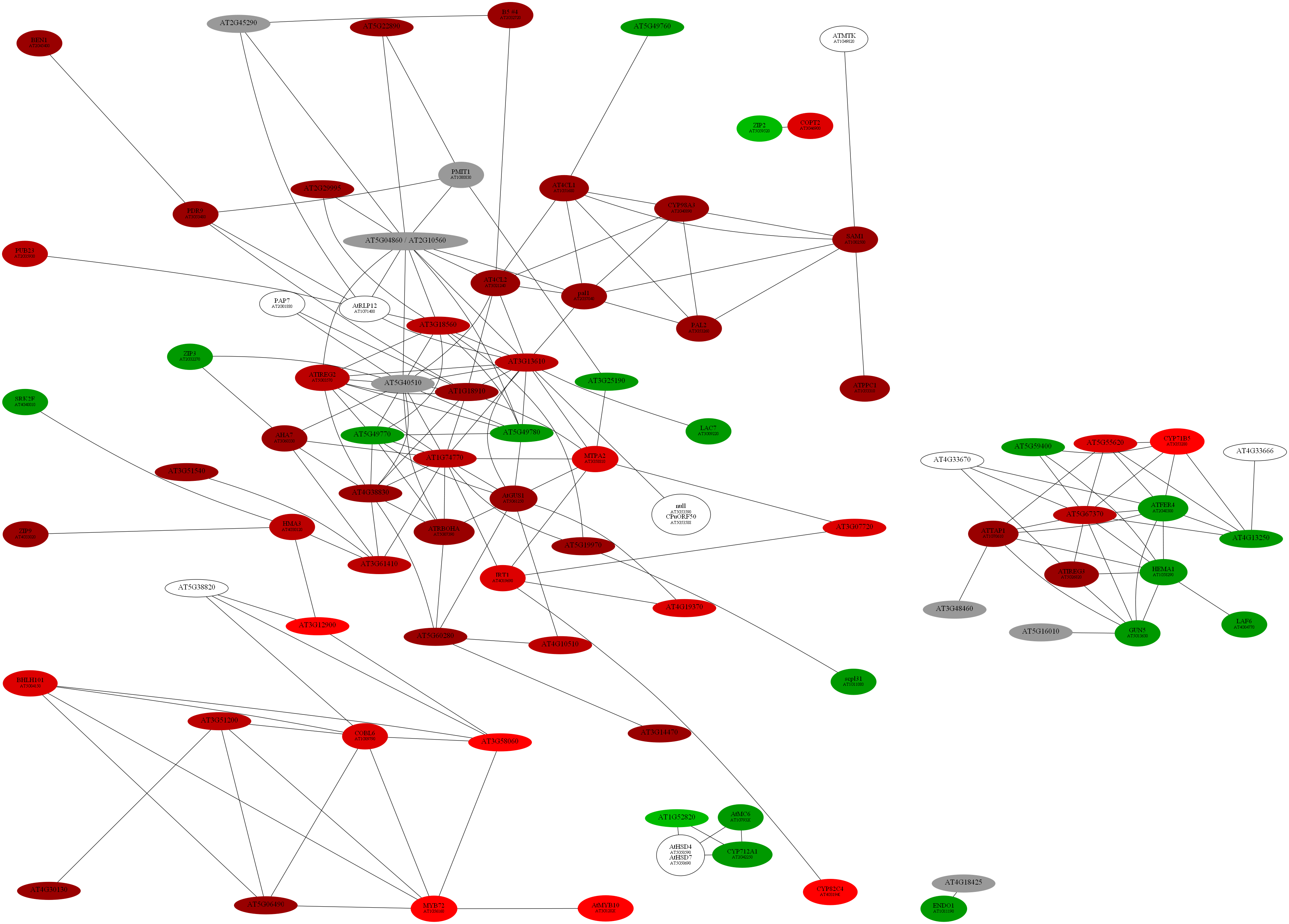
D:\wdlin\Project1\maccu\data\20091203_example>java -Xmx1400M -classpath maccu.jar maccu.GraphAdjust -I root.graph -O rootRemove -remove 1436.graph
program: GraphAdjust
input graph (-I): root.graph
output prefix (-O): rootRemove
remove node list (-RN): []
remove edge list (-RE): []
remove graph list (-remove): [1436.graph]
retain graph list (-retain): []
degree filter (-D): 0
cluster-size filter (-S): 1
cluster 0, size: 56
cluster 1, size: 15
cluster 2, size: 4
cluster 3, size: 2
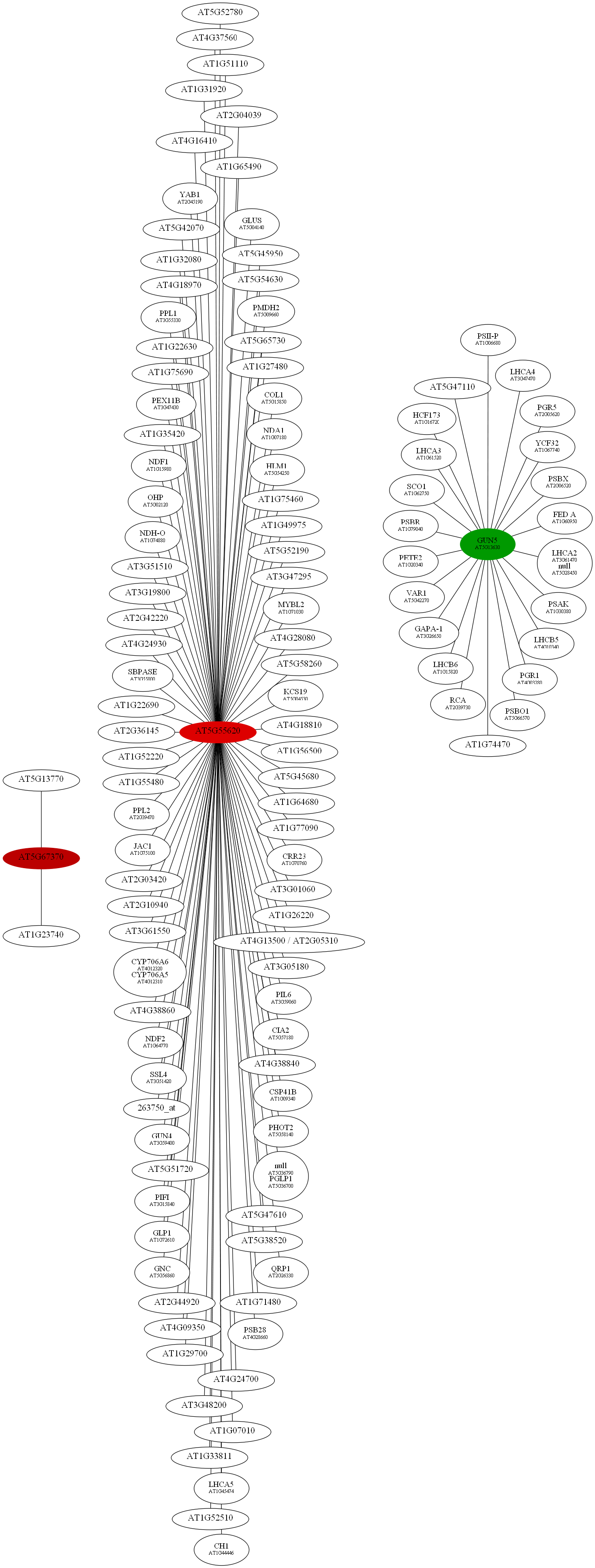
D:\wdlin\Project1\maccu\data\20091203_example>java -classpath maccu.jar maccu.GraphMakeUp -I rootRemove-ALL.graph -O rootRemove.dot -label geneSymbols.txt -fold colorALL.txt
program: GraphMakeUp
input graph (-I): rootRemove-ALL.graph
output filename (-O): rootRemove.dot
label filename (-label): geneSymbols.txt
fold change filename (-fold): colorALL.txt
coloring list (-C): {}
font 1 (-font1): 12
font 2 (-font2): 8
D:\wdlin\Project1\maccu\data\20091203_example>twopi -Goutputorder=edgesfirst -Goverlap=vpsc -Granksep=2 -Gratio=auto -Tpng -o rootRemove.png rootRemove.dot
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project1\maccu\data\20091203_example>java -Xmx1400M -classpath maccu.jar maccu.CoExpressFishing -I ATH1_Root_RMA.tdt -C ATH1_Root_RMA.correl -O fishing -B root-1.graph -assign ATH1_ProbeLocusMap.TXT -CF 0.9 -STEP 1 -BUFFER false
program: CoExpressFishing
expression filename (-I): ATH1_Root_RMA.tdt
probe assignment filename (-assign): ATH1_ProbeLocusMap.TXT
correlation filename (-C): ATH1_Root_RMA.correl
output filename (-O): fishing
bait filename (in locus) (-B): root-1.graph
step limit (-STEP): 1
buffer edge (-BUFFER): false
number filer (-NF): 8
correl filer (-CF): 0.9
reading expression
reading bait
computing edges
edge computeing, dist = 0, buffer size = 15
reach step limit.
graph building
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project1\maccu\data\20091203_example>java -classpath maccu.jar maccu.GraphMakeUp -I fishing.graph -O fishing.dot -label geneSymbols.txt -fold colorALL.txt
program: GraphMakeUp
input graph (-I): fishing.graph
output filename (-O): fishing.dot
label filename (-label): geneSymbols.txt
fold change filename (-fold): colorALL.txt
coloring list (-C): {}
font 1 (-font1): 12
font 2 (-font2): 8
D:\wdlin\Project1\maccu\data\20091203_example>twopi -Goutputorder=edgesfirst -Goverlap=vpsc -Granksep=2 -Gratio=auto -Tpng -o fishing.png fishing.dot
D:\wdlin\Project1\maccu\data\20091203_example>
D:\wdlin\Project0\gobu>java -Xmx600M -classpath gobu.jar bio301.goutil.gobu.data.AddAnnotation -I d:\wdlin\Project1\maccu\data\20091203_example\root.tree -S d:\wdlin\Project1\maccu\data\common\LocusAnnotation.list -O d:\wdlin\Project1\maccu\data\20091203_example\root.raw.tree -OBO data\gene_ontology.1_2.obo
program: AddAnnotation
input filename (-I): d:\wdlin\Project1\maccu\data\20091203_example\root.tree
subtree filename (-S): d:\wdlin\Project1\maccu\data\common\LocusAnnotation.list
output filename (-O): d:\wdlin\Project1\maccu\data\20091203_example\root.raw.tree
OBO filename (-OBO): data\gene_ontology.1_2.obo
coverage check (-C): false
D:\wdlin\Project0\gobu>java -Xmx600M -classpath gobu.jar bio301.goutil.gobu.data.TreeSorter -I d:\wdlin\Project1\maccu\data\20091203_example\root.raw.tree -O d:\wdlin\Project1\maccu\data\20091203_example\root.sort.tree -OBO data\gene_ontology.1_2.obo
program: TreeSorter
input filename (-I): d:\wdlin\Project1\maccu\data\20091203_example\root.raw.tree
output filename (-O): d:\wdlin\Project1\maccu\data\20091203_example\root.sort.tree
OBO filename (-OBO): data\gene_ontology.1_2.obo
D:\wdlin\Project0\gobu>
d:\wdlin\Project0\gobu>gobuCmd.bat
d:\wdlin\Project0\gobu>java -Xmx900M -classpath gobu.jar bio301.goutil.gobu.GobuFrame
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}